What is SDH-deficient gastrointestinal stromal tumor?
published by the National Cancer Institute
SDH-deficient gastrointestinal stromal tumor, or SDH-deficient GIST, is a type of digestive tract cancer. It almost always grows in the stomach or small intestine, but sometimes in the large
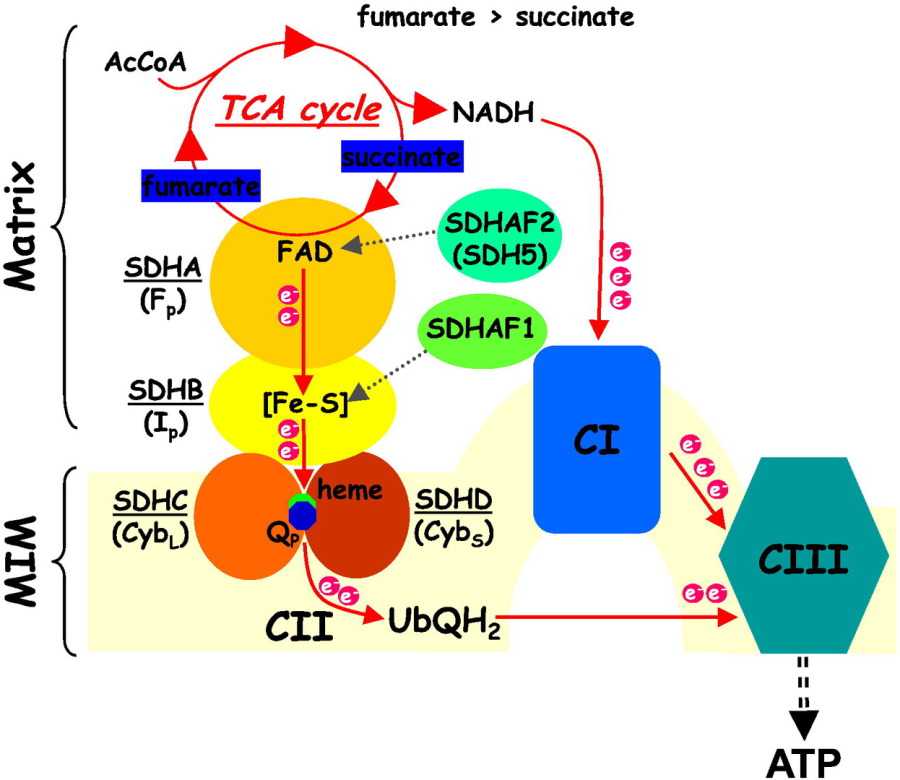
intestine. GISTs are “SDH-deficient” 5% to 7.5% of the time and usually grow only in the stomach. SDH is short for succinate dehydrogenase, a protein that is important for making energy for your
body. Most people with SDH-deficient GISTs are children and some young adults.
People with SDH-deficient GIST are at higher risk for other types of cancer. They are at risk for pulmonary chondromas, a benign tumor in the lung, and paragangliomas, a rare tumor that grows in the
nerve cell and can occur anywhere in the body.
How common is SDH-deficient gastrointestinal stromal tumor?
GIST is a common type of tumor of the digestive tract. It is thought that 4,000 to 6,000 people per year are diagnosed with GIST in the United States. SDH-deficient GIST is much rarer, making up 5%
to 7.5% of all GISTs. SDH-deficient GIST affects females more than males.
How is SDH-deficient gastrointestinal stromal tumor diagnosed?
Symptoms of SDH-deficient GIST include bleeding in the digestive tract, intense abdominal pain, feeling tired, or feeling full after a small meal. For many people, it can be a difficult and
frustrating process to get the correct diagnosis. GIST is hard to diagnose because it looks like other types of stomach cancer.
Imaging: If you have symptoms of SDH-deficient GIST, your doctor will use imaging scans such as CT or MRI to look at the size of the tumor and where it is in your digestive tract. Your
doctor may also use endoscopy to look at the tumor. Endoscopy is a type of imaging where a small video camera on a long flexible tube is put inside your digestive tract.
Biopsy: To check if the tumor is SDH-deficient GIST, your doctor will perform a biopsy, taking a small sample from the tumor with a needle. A pathologist will study cells from the
sample under the microscope to see what kind of tumor it is. This may be done at the same time as the endoscopy.
Genetic testing: Your doctor will use the biopsy to check the SDH genes
in your tumor. This will indicate whether you have SDH-deficient GIST or another type.
How is SDH-deficient gastrointestinal stromal tumor treated?
SDH-deficient GIST can be taken out with surgery. It does not respond well to some chemotherapy, the way other GISTs do, but new chemotherapy options are being studied that have shown promise.
Does SDH-deficient gastrointestinal stromal tumor run in families?
Yes. In rare cases, SDH-deficient GISTs can be passed down in families. Carney-Stratakis syndrome is a disease where people develop several types of tumors including SDH-deficient GIST. Many of the
families with Carney-Stratakis syndrome carry mutations in SDH that can be passed on to children.
How does SDH-deficient gastrointestinal stromal tumor form?
Scientists are always working to understand how cancer forms but each form can be hard to prove. We know that SDH can be turned off in many different ways in SDH-deficient GIST. When SDH is turned
off, cells can become overactive and behave as if your body is growing or repairing a wound. Scientists are trying to understand how tumors form when cells act in this way.
What is the prognosis for someone with SDH-deficient gastrointestinal stromal tumor?
The estimate of how a disease will affect you long-term is called prognosis. Every person is different and prognosis will depend on many factors, such as:
-
Where the tumor is in your body
-
If the cancer has spread to other parts of your body
-
How much of the tumor was taken out during surgery
If you want information on your prognosis, it is important to talk to your doctor. NCI also has resources
to help you understand cancer prognosis.
Doctors estimate survival rates by how groups of people with SDH-deficient GIST have done in the past. Because there are so few people with SDH-deficient GIST, these rates may not be very accurate.
They also can’t consider newer treatments being developed.
People can live with SDH-deficient GIST for a very long time, especially if the tumor is taken out in surgery and doesn’t spread to other parts of the body.
For More Information
NIH Pediatric and Wildtype GIST Clinic
Each year, NIH staff host the Pediatric and Wildtype GIST Clinic at the NIH Clinical Center. The clinic is a collaborative effort between researchers, doctors, and advocates with the goal of helping
young patients with GIST. The 2020 GIST Clinic will be held June 10-12, 2020. Visit
the clinic website to register and learn more.